Lowell’s melt inclusion workflow
Rev. LRM 9/10/2021 -- original version
Rev. LRM 12/15/2021 -- updated and adapted for this blog
0) Forward
This document is intended to compensate for inadequate sample preparation descriptions in the “methods” sections of various papers about melt inclusions. In the interest of time, this document was written almost entirely from memory, so some of the information may be incorrect. In other words, it’s about as good as talking to me over a beer in a poster hall. Also note that this workflow assumes that the researcher is working on a project that is similar in scope to my research: using melt inclusions from basaltic volcanoes to determine the pre-eruptive volatile content of the magma, and maybe of the melting source material. There is no “universal” method for preparing melt inclusions, but these instructions are probably better than nothing at all!
1) Bulk rock processing
Crush whole rock material so that inclusion-bearing phenocrysts can be isolated from unwanted (matrix) material. If samples were collected as lavas, use a rock saw to cut the rock into cm-scale pieces, and crush them in a benchtop jaw crusher (a big hammer also works). If the samples were collected as tephras (e.g. ash, lapilli), then a big mortar and pestle may also be used. After crushing, wash the crushed material to remove the dust using water; any water is fine, it doesn’t have to be distilled, deionized, etc. You’re not going to find any useful host phenocrysts in the very fine material.
2) Mineral separation
a) Sieving
It’s a good idea to sieve the sample material into several different fractions; the exact sieve sizes don’t matter. Particlesabove a certain size (e.g. > a few mm) willcomprise host phenocrysts plus additional matrix material. Particles below a certain size (e.g. <100s of microns) will only contain fragments of host phenocrysts. The ideal particle size, somewhere in between, will contain whole phenocrysts with minimal adhering matrix. I have had a lot of luck finding nice, intact olivine phenocrysts with US standard US mesh sizes 60 and 35 (250 micron and 500 micron, respectively).
b) Picking
Olivines in crushed lava samples can be easily hand-picked under a low-powered binocular microscope with tweezers. Olivines from fresh tephra samples can be more difficult to find without the use of transmitted cross-polarized light. Place the crushed rock sample in a transparent glass dish with a transmitted light source below the sample, one polarizing film between the light and the sample, and one polarizing film above the sample. When the polarizing films are oriented correctly, the glass (isotropic) will go extinct and the olivines (biaxial) will appear illuminated. Sometimes this helps with sufficiently fine-grained lava samples too.
3) Host mineral selection
a) Epoxy mount preparation
Place olivines on double-sided tape surrounded by a 1-inch (inner diameter) circular mold. Pour epoxy over the olivines. I like to use the single use green “double bubble” brand epoxy packets, but any clear epoxy is probably ok. Once the epoxy has cured, polish the grain mount until the interior of a sufficient number of the phenocrysts can be seen.
b) Selection considerations
I generally look for melt inclusions which are glassy and at least 25 microns in diameter –this corresponds roughly to the raster area in SIMS analysis.Avoid melt inclusions which are intersected by cracks before polishing. Sometimes cracks might form around melt inclusions during polishing but that is not necessarily a problem. Crystals containing suitable melt inclusions can be removed using a soldering iron to soften the epoxy surrounding the crystal and tweezers to pick the crystal out.
4) Melt inclusion preparation
a) Single grain polishing
Note: to give proper credit, I learned this method from some of Paul Wallace’s students.
Note:I recommend frequently examining the sample under the microscope and taking pictures of the inclusions before they are exposed. Detailed photographs of melt inclusions are important 1) because cracking and other damage can occur during the polishing process, and 2) because samples can be easily reidentified in the event that they fall on the floor or become mixed up. You never know which photos will be important later, so try to take journal-quality photos every time!
For this process, I like to use CrystalBond 590 mounting adhesive to attach single crystals onto 1-inch round glass slides. CrystalBond is a hard resin that is purchased in a ~1 inch round cylinder. When heated to ~50C, the resin softens and can be used like epoxy; when cooled back to room temperature, it hardens instantly. To use it, you break off a small piece usingpliers or a hammer, place the piece on a glass slide, put the slide on a hot surface such as a hot plate, attach the crystal using tweezers, and remove the sample from the heat. Instead of a hot plate, I prefer to use a thermoelectric plate (a.k.a. a Peltier plate) with an adjustable benchtop power supply. This method has several advantages over a hot plate: 1) it takes up less space, so it’s easier to do under a microscope and allows better control of the sample, 2) you’re much less likely to burn yourself, and 3) this equipment can also be used to heat samples during Raman analysis (see below).
b) Polishing
Here are the polishing steps which have worked for me:
- 800 grit SiC paper on a wet glass plate for 0.5 to 5 minutes or until the inclusion is ~10 microns below the surface.
- 3 micron diamond lapping film at ~150 rpm on a polishing wheel for 30 to 90 seconds or until the glass is exposed(without breaching the bubble!). Sometimes it’s best to stop and do Raman analyses before polishing into the glassy part of the melt inclusion if the bubble appears to be very close to the polished-side of the inclusion.
- 0.3 micron alumina powder and fuzzy silk pad. In our lab, the silk pad is usually attached to a round plate which is used on the polishing wheel. I just polish for ~30 seconds by hand without turning on the wheel. The goal of this step is to make the surface of the sample perfectly smooth. You know it’s smooth enough if you can’t find the surface in reflected light at 100x magnification.
Note: Here is a video I made to demonstrate the single crystal polishing process. Apologies for the poor quality! https://www.youtube.com/watch?v=QhwF81MxSTo&authuser=0
Note: Here is another video about a peltier plate setup I use to heat and cool samples for sample preparation and Raman analysis: https://www.youtube.com/watch?v=V8zZxnxeHVg&authuser=0
c) Sample storage and organization
For compact storage and to reduce the cost of round glass slides, I prefer to store crystal samples on a petrographic slide sitting in the drop of CrystalBond used during polishing. To remove the sample from the round glass slide, heat the slide just a little bit (maybe ~40-50C) and then use a razor blade to cut the Crystalbond free from the glass. Then heat the petrographic slide and stick the sample on. Again, this isn’t totally necessary, and you can also just store your samples on a bunch of individual round glass slides.
5) Raman analysis
Note: melt inclusion bubbles aren’t always empty. This observation has been documented thoroughly at the time of writing. For example, the image below shows a melt inclusion from Maui (~50 micron diameter) with a bubble containing a mixture of liquid + vapor. The changing light level in the image occurs because I am inserting/removing an infrared filter placed between the transmitted light source and the sample.
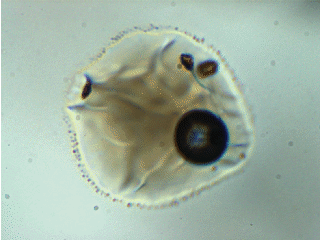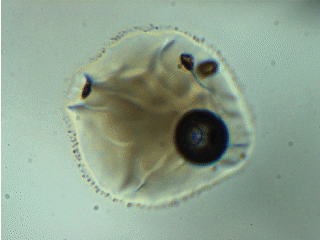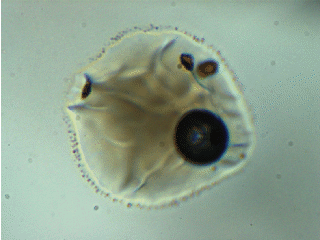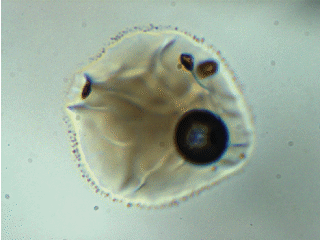
To account for CO2 contained in bubbles, I analyze them using Raman spectroscopy and use an empirical relationship between the separation of the Fermi diad and fluid densityto calculate the mass of CO2 in the bubble.
a) Hardware considerations
Many labs have Raman spectrometers, but they are not all appropriate for analyzing fluid and melt inclusions. Before doing Raman analyses, I recommend requesting that the lab provide an example spectrum showing the “Fermi diad.” The spectrum should have a resolution of 0.5 cm-1 or better. In other words, open the spectrum as a text file, and each point on the x-axis should be within 0.5 wavenumbers of the next. The Raman in our lab has an 800 mm focal length, and we use a 1800 mm-1 diffraction grating when analyzing CO2. This relatively long focal length means that the diffracted “rainbow” of light can spread out sufficiently between the diffraction grating and the detector, which provides the necessary spectral resolution. Additionally, a Raman system should have well-aligned optics -- this is necessary for a good signal/noise ratio. A simple way to evaluate the signal quality when analyzing CO2 is to count the number of points in each CO2 peak. If by visual inspection one or both of the peaks are comprised of fewer than two points above background, then the signal is too poor to use. If both peaks are comprised of more than 3-5 points above background, then the spectrum is probably adequate. If you can see the so-called “hot bands” on either side of the “Fermi diad,” then you have a great signal!
b) The problem with high-density CO2
Suppose you’re analyzing a fluid on the Raman at room temperature, and you calculate a density of ~0.4g/cc. This presents a potential problem because bulk densities in the range of roughly 0.2 to 0.7g/cc represent the interval over which CO2 exists as a mixture of liquid and vapor. That means that the Raman spectrum probably contains two sets of overlapping fermi diad peaks,and the magnitude of each could potentially depend on where the laser is focused within the sample. In the past, I have approached this problem by heating the sample to 35C using my thermoelectric plate setup while the sample is being analyzed on the Raman spectrometer. To monitor the temperature, I use a thermocouple which is placed as close to the host crystal as possible. It also helps to use a thermoelectric plate with a small hole in the center, which allows the sample to be viewed in transmitted light. In theory, heating the sample above the critical point for CO2 in this way should homogenize the fluid so that the composition of the bubble becomes homogeneous. In practice, I’m not sure whether laser heating would homogenize the bubble anyway. If this is the case, then actively heating samples as I have done is probably unnecessary. Somebody should figure this out if it has not already been done.
6) Secondary ion mass spectrometric (SIMS) analysis
a) Preparing Indium mounts
Samples analyzed using SIMS must be mounted in indium to prevent outgassing of volatiles released from epoxy/resin under vacuum. I’m not sure this is necessary for all samples –- it may only apply to those being analyzed for volatile elements at low concentrations. That’s a question for your SIMS lab manager. I have prepared all ofmy indium mounts using 1-inch aluminum molds which were provided by the nice people at the Woods Hole Oceanographic Institute. I also used a couple of additional custom parts machined in my department: a ~1 inch diameter steel cylinder for pressing the surface of the indium to be smooth, and a >1 inch diameter aluminum receptacle to allow indium to flow out from holes in the back of the mold. I used high-purity indium “shot” purchased from a lab supplier to fill the mount.
Here are the stepsto make the indium mount:
- Place the aluminum mold on a hot plate, and heat it above the melting point for indium. I think it’s around 250C or so.
- Add enough indium to fill the mold, and wait for it to melt.After the indiumhas melted, it may be necessary to add a little more indium to bring the level of molten indium above the edge of the mold. The indium should be cohesive enough that it stays in the mold without wetting the aluminum and flowing out.
- Turn off the hot plate, and wait for the mold to cool enough to handle.
- Place themold between the steel cylinderand the extra aluminum receptacle, and then place all of these into the jaws of a benchtop vise. Tighten the vise firmly –- the desired result is that the top surface of the mold will be flat, and the excess indium will flow out of the little holes in the back of the mold.
- Trim out the excess indium with a razor blade, and press the mount a few more times as necessary to ensure that the top surface of the mount is perfectly flat.
After Raman analysis, my samples are still surrounded by Crystalbond, so these are the steps I use to clean the crystals:
Note: I prefer to do all of the steps involving acetone in a fume hood. I’m told that acetone fumes are unhealthy to breathe.
- Heat the sample to soften the CrystalBond, and remove as much of it as possible using tweezers and/or a razor blade.
- Transfer the crystal into asmall glass containerfilled with acetone, and cover using parafilm. I use a ~1 cm diameter x ~3 cm tall straight-walled glass vial.
- Place the vial in an ultrasonic bath for ~3 minutes-Pour out the crystal + acetone into a funnel lined with a Kimwipe, and hang the Kimwipe to dry for about 3 minutes.
- Retrieve the crystal from the Kimwipe, and inspect under the microscope for epoxy/Crystalbond residue.
Note: I don’t remember exactly how this worked, but I remember sonicating each crystal in acetone twice. I would always have three crystals cleaning at the same time where Crystal A would be drying, Crystal B would be sonicating in clean water, and Crystal C would be sonicating in the recycled acetonefrom Crystal B. When the crystals are cleaned, press them into the indium mount with the polished side and exposed melt inclusions facing outward. I prefer to use a wooden popsicle stick for this.
b) Lessons learned on the SIMS
In my experience, samples are always poorly illuminated and viewed using a terrible potato-quality camera during SIMS analysis. If samples are rounded, melt inclusions may be lost in shadow. If the surfacesof the exposed melt inclusions are flush with the host mineral, they may be impossible to find. For these reasons, I recommend making detailed maps of the indium mount with many photographs taken in reflected light. Cracks and grain boundaries will serve as useful landmarks when trying to locate exposed melt inclusions on the SIMS. If you have access to a laser ablation system, you might even consider drilling a few holes in the sample with the laser to mark the locations of melt inclusions. All of the times that I used a SIMS, the samples were gently cleaned usingvarious simple methods (alcohol + Kimwipe or compressed air + deionized water) and then Au-coated at the facility where the analyses were done.
7) Electron probe microanalysis (EPMA)
a) EPMA sample preparation
In the past, I have always done my microprobe analyses with carbon-coated melt inclusions based on carbon-coated micro beam standards. For this reason, I prefer to do microprobe analyses after SIMS analysesto avoid the potential for carbon contamination. This can be problematic though because thetopography of the SIMS crater may affect the quality of the microprobe analyses.To preparethe samples for EPMA, gently polish the indium mount by hand using 0.3micron alumina powder and a fuzzy silk polishing padto remove Au coating from SIMS analyses.Removing all of the coating is basically impossible, but you should at least be able to remove enough to expose aclean, approximately 5 micronwide spot for microprobe analyses
8) Laser ablation analyses
Do we really have to do this? Can’t we just use trace element data from whole rocks? Is the trace element heterogeneity observed in melt inclusions actually related to magma evolution, or is it just an artifact of various post-entrapment processes that nobody really cares about? Is it really necessary that each melt inclusion has a complete geochemical analysis rather than just comparing major, trace, and volatile elements using summary statistics? I’ll let you be the judge.
Laser ablation comes with two major difficulties: 1) the availability of transmitted light, and 2) the availability of a large analytical area. Because I wanted to use the largest beam diameter possible and make sure that the beam was positioned accurately, I decided to transfer my crystals from the SIMS indium mount back into a clear epoxy mount. To extract the crystals from the indium mount, I just used a razor blade to open up some space on the side of each crystal and tweezers to work each crystal out. Removing the first crystal from the indium is tricky, but it was easier for the subsequent crystals.This sounds crazy, but it wasn’t as difficult as I expected, and the sample loss ratewas probably less than10%.During the laser ablation analyses, I positioned the laser at the center of each inclusion as viewed in transmitted light so that the spot would fall well within the boundary of the inclusion allowing for the inclusion to be tilted or irregularly shaped. This method yielded decent-looking plateaus most of the time.
For me, laser ablation analysis seems to have many opportunities for method development, but I did not have time to explore these before I finished my research projects. In retrospect, I wish I could have spent more time improving my analytical strategy and learning about the (very complicated!) data reduction process.